Paediatric orthopaedics
Bone basics
Normal bone may be either cortical or cancellous, both of which have a highly ordered macrostructure leading to the ability to withstand stresses. Mature bone is stress orientated, allowing it to resist forces in some directions but being weaker to other force directions. Immature bone is woven and has a less organised structure, leading to it being weaker and more flexible. Bone is made up of a matrix containing organic components
(type I collagen, proteins, proteoglycans and growth factors) that provide some strength but also promotes bone mineralisation and cell activity; inorganic components (calcium hydroxyapatite and calcium phosphate) which provide compressive strength; and bone cells (osteoBlasts - BUILD new bone; osteoClasts - CHEW up/digest bone; osteocytes - maintain bone).
Bone growth
Long bones undergo longitudinal growth at the ends via the growth plate (physis). This is via a process called enchondral bone formation in which cells form cartilage that becomes mineralised. This is then replaced by bone when osteoclasts reabsorb it, closely followed by osteoblasts laying down new bone. Bone that has grown at the physis matures at the metaphysis and as the epiphysis grows further away, it will become the shaft of the long bone (diaphysis). The joint is formed by growth and development of special cartilage at the epiphysis.
Flat bones are formed via a process called intramembranous bone formation in which mesenchymal cells aggregate and differentiate into osteoblasts and deposit an organic matrix that mineralises into bone. There is no cartilage precursor in intramembranous bone formation.
Lower limb development
From birth through to full skeletal maturity, a child’s lower limbs will change in alignment as they grow. When born, until up to 18 months of age, children will have genu varum (bowed legs). This progresses to genu valgum by 3 years of age and gradually settles to a physiological mild valgus by age 5 years. If the genu varum does not resolve by 2 years of age, the problem is unilateral or there is severe angulation, then a pathological cause must be considered, such as Blount’s disease (growth disorder of the proximal medial tibia resulting in genu varum and internal tibial torsion).
As children start walking, parents may notice an in-toeing gait. The most common cause is internal rotation of the tibia (internal tibial torsion) due to intrauterine moulding that virtually always resolves spontaneously with growth. Similarly, children are born with their proximal femora naturally internally rotated due to their intrauterine position. This can also lead to an in-toed gait and sitting in a W position with legs behind them. This usually corrects spontaneously by adolescence and rarely requires treatment.
Many young children, when first starting to walk, will walk on tip-toes or with flat feet. Habitual toe walking may be due to contracture of the Achilles tendon and usually responds to stretching but occasionally requires serial casting or even Achilles lengthening. Flat foot deformity is seen when standing and is often associated with ligamentous laxity. Medial arch supports have not been shown to result in any correction in deformity and the majority of flat feet do not cause any disability.
During growth, stress from the patellar tendon upon the tibial tubercle can lead to fatigue failure, leading to pain and tenderness with localised swelling over the tubercle. This is termed Osgood-Schlatter’s disease and is a self-limiting phenomenon only requiring activity modification when symptoms are present, usually during teenage growth spurts. A similar phenomenon is seen in the Achilles tendon leading to pain in the posterior heel (calcaneum) around the adolescent growth spurt and is known as Sever’s disease.
Normal bone may be either cortical or cancellous, both of which have a highly ordered macrostructure leading to the ability to withstand stresses. Mature bone is stress orientated, allowing it to resist forces in some directions but being weaker to other force directions. Immature bone is woven and has a less organised structure, leading to it being weaker and more flexible. Bone is made up of a matrix containing organic components
(type I collagen, proteins, proteoglycans and growth factors) that provide some strength but also promotes bone mineralisation and cell activity; inorganic components (calcium hydroxyapatite and calcium phosphate) which provide compressive strength; and bone cells (osteoBlasts - BUILD new bone; osteoClasts - CHEW up/digest bone; osteocytes - maintain bone).
Bone growth
Long bones undergo longitudinal growth at the ends via the growth plate (physis). This is via a process called enchondral bone formation in which cells form cartilage that becomes mineralised. This is then replaced by bone when osteoclasts reabsorb it, closely followed by osteoblasts laying down new bone. Bone that has grown at the physis matures at the metaphysis and as the epiphysis grows further away, it will become the shaft of the long bone (diaphysis). The joint is formed by growth and development of special cartilage at the epiphysis.
Flat bones are formed via a process called intramembranous bone formation in which mesenchymal cells aggregate and differentiate into osteoblasts and deposit an organic matrix that mineralises into bone. There is no cartilage precursor in intramembranous bone formation.
Lower limb development
From birth through to full skeletal maturity, a child’s lower limbs will change in alignment as they grow. When born, until up to 18 months of age, children will have genu varum (bowed legs). This progresses to genu valgum by 3 years of age and gradually settles to a physiological mild valgus by age 5 years. If the genu varum does not resolve by 2 years of age, the problem is unilateral or there is severe angulation, then a pathological cause must be considered, such as Blount’s disease (growth disorder of the proximal medial tibia resulting in genu varum and internal tibial torsion).
As children start walking, parents may notice an in-toeing gait. The most common cause is internal rotation of the tibia (internal tibial torsion) due to intrauterine moulding that virtually always resolves spontaneously with growth. Similarly, children are born with their proximal femora naturally internally rotated due to their intrauterine position. This can also lead to an in-toed gait and sitting in a W position with legs behind them. This usually corrects spontaneously by adolescence and rarely requires treatment.
Many young children, when first starting to walk, will walk on tip-toes or with flat feet. Habitual toe walking may be due to contracture of the Achilles tendon and usually responds to stretching but occasionally requires serial casting or even Achilles lengthening. Flat foot deformity is seen when standing and is often associated with ligamentous laxity. Medial arch supports have not been shown to result in any correction in deformity and the majority of flat feet do not cause any disability.
During growth, stress from the patellar tendon upon the tibial tubercle can lead to fatigue failure, leading to pain and tenderness with localised swelling over the tubercle. This is termed Osgood-Schlatter’s disease and is a self-limiting phenomenon only requiring activity modification when symptoms are present, usually during teenage growth spurts. A similar phenomenon is seen in the Achilles tendon leading to pain in the posterior heel (calcaneum) around the adolescent growth spurt and is known as Sever’s disease.
Disorders of bone growth
Bone dysplasias (dwarfism)
These affect specific areas of the growing bone and have varying affects on different bones within the body. Involved bones are typically shortened - if all bones are affected equally, this is proportionate dwarfism. If there is greater involvement of the spine, resulting in a short trunk, or the limbs, resulting in short limbs, this is know as disproportionate dwarfism. Achondroplasia is an autosomal dominant condition and the most common form of dwarfism resulting in a disproportionate child with short limbs. Other examples of disproportionate dwarfs include mulitple ephiphyseal dysplasia, diastrophic dysplasia (both short limb) and spondyloepiphyseal dysplasia (short trunk). Examples of proportionate dwarfisms include the mucopolysaccharidoses (Morquio’s, Hurler’s, Hunter’s and Sanfilippo’s syndromes). By the way, this is a very specialized area of study and
knowledge of these syndromes is not expected at undergraduate level!
Osteogenesis Imperfecta (brittle bone disease)
A defect in volume and quality of type I collagen, leading to bone fragility, short stature, tooth defects, ligamentous laxity and hearing defects. Treatment is management of acute fractures and prevention of deformity and further fractures. Bracing may be required to prevent deformity although this is commonly not successful for scoliosis. Fractures are often managed non-operatively or with minimal internal fixation and will heal well.
Osteopetrosis (marble bone disease)
Decreased osteoclast function leads to bony sclerosis and thickening with loss of the bone medullary canal. The bone is incredibly hard but can be prone to fracture because it is abnormally brittle.
Rickets
A decrease in calcium and occasionally phosphorous which affects the mineralisation occurring at the epiphyses of long bones. There are many different causes of rickets including dietary insufficiency, gastrointestinal malabsorption, kidney disease and genetic abnormalities of enzymes. Rickets leads to weak, brittle bones, bowing of the long bones, flattening of the skull, kyphosis and enlargement of the costal cartilages (rickety rosary).
Radiographic examination reveals widening and cupping of the physes of long bones with marked bowing, particularly of the femur and tibia.
Treatment is according to the underlying cause, often requiring administration of calcium, phosphate and/or Vitamin D or one of its more active metabolites.
Bone dysplasias (dwarfism)
These affect specific areas of the growing bone and have varying affects on different bones within the body. Involved bones are typically shortened - if all bones are affected equally, this is proportionate dwarfism. If there is greater involvement of the spine, resulting in a short trunk, or the limbs, resulting in short limbs, this is know as disproportionate dwarfism. Achondroplasia is an autosomal dominant condition and the most common form of dwarfism resulting in a disproportionate child with short limbs. Other examples of disproportionate dwarfs include mulitple ephiphyseal dysplasia, diastrophic dysplasia (both short limb) and spondyloepiphyseal dysplasia (short trunk). Examples of proportionate dwarfisms include the mucopolysaccharidoses (Morquio’s, Hurler’s, Hunter’s and Sanfilippo’s syndromes). By the way, this is a very specialized area of study and
knowledge of these syndromes is not expected at undergraduate level!
Osteogenesis Imperfecta (brittle bone disease)
A defect in volume and quality of type I collagen, leading to bone fragility, short stature, tooth defects, ligamentous laxity and hearing defects. Treatment is management of acute fractures and prevention of deformity and further fractures. Bracing may be required to prevent deformity although this is commonly not successful for scoliosis. Fractures are often managed non-operatively or with minimal internal fixation and will heal well.
Osteopetrosis (marble bone disease)
Decreased osteoclast function leads to bony sclerosis and thickening with loss of the bone medullary canal. The bone is incredibly hard but can be prone to fracture because it is abnormally brittle.
Rickets
A decrease in calcium and occasionally phosphorous which affects the mineralisation occurring at the epiphyses of long bones. There are many different causes of rickets including dietary insufficiency, gastrointestinal malabsorption, kidney disease and genetic abnormalities of enzymes. Rickets leads to weak, brittle bones, bowing of the long bones, flattening of the skull, kyphosis and enlargement of the costal cartilages (rickety rosary).
Radiographic examination reveals widening and cupping of the physes of long bones with marked bowing, particularly of the femur and tibia.
Treatment is according to the underlying cause, often requiring administration of calcium, phosphate and/or Vitamin D or one of its more active metabolites.
Cerebral palsy (CP)
CP is a non-progressive disorder, resulting from injury to the immature brain, leading to an upper motor neurone disorder of the neuromuscular system with age of onset less than 2 years of age. It may be caused by intrauterine, perinatal and postnatal problems, including infection, prematurity, anoxia and meningitis. It results in muscle weakness and spasticity which initially cause tightness around the joints, progressing to fixed deformity and contractures. This can eventually lead to bone deformity and joint subluxation or dislocation (typically of the hip). CP may involve different parts of the body, including scoliosis of the spine, and results in hemiplegia, diplegia or total body involvement.
Management involves a multi-disciplinary team involving paediatricians, teachers, physiotherapists, orthotists and surgeons. Treatment may be required to improve function by preventing development of contractures and deformity using splints, stretching exercises and injections (often using selective Botulinum toxin to paralyse spastic muscles).
Abnormalities of gait may be documented using special gait analysis labs and patients may require tendon lengthening and transfers to improve their gait.
Scoliosis is common and may require operative correction and spinal fusion if bracing and custom moulded wheel-chair seats fail.
Hip subluxation and dislocation may initially be treated with tendon releases (often the adductor and psoas tendons are the major deforming forces and require lengthening or release) but may require femoral or acetabular osteotomies to improve stability.
CP is a non-progressive disorder, resulting from injury to the immature brain, leading to an upper motor neurone disorder of the neuromuscular system with age of onset less than 2 years of age. It may be caused by intrauterine, perinatal and postnatal problems, including infection, prematurity, anoxia and meningitis. It results in muscle weakness and spasticity which initially cause tightness around the joints, progressing to fixed deformity and contractures. This can eventually lead to bone deformity and joint subluxation or dislocation (typically of the hip). CP may involve different parts of the body, including scoliosis of the spine, and results in hemiplegia, diplegia or total body involvement.
Management involves a multi-disciplinary team involving paediatricians, teachers, physiotherapists, orthotists and surgeons. Treatment may be required to improve function by preventing development of contractures and deformity using splints, stretching exercises and injections (often using selective Botulinum toxin to paralyse spastic muscles).
Abnormalities of gait may be documented using special gait analysis labs and patients may require tendon lengthening and transfers to improve their gait.
Scoliosis is common and may require operative correction and spinal fusion if bracing and custom moulded wheel-chair seats fail.
Hip subluxation and dislocation may initially be treated with tendon releases (often the adductor and psoas tendons are the major deforming forces and require lengthening or release) but may require femoral or acetabular osteotomies to improve stability.
Paediatric hip disorders
Developmental dysplasia of the hip (DDH)
DH is caused by abnormal development of the acetabulum and proximal femur leading to subluxation or dislocation of the hip. It is usually noted at birth during the new born check with the Ortolani and Barlow tests that will reveal a clicking, dislocated or unstable hip. The incidence is 1 in 1000. There should be a high index of suspicion in patients with risk factors such as family history, breech position, female sex and oligohydramnios, and
should be followed up with specific hip examination and ultrasound scanning must be performed. Clinical examination may show positive Galeazzi test (affected thigh is shorter when both legs are held together with knees flexed) and decreased abduction on the dislocated side. Asymmetrical skin folds are a non-specific sign.
Developmental dysplasia of the hip (DDH)
DH is caused by abnormal development of the acetabulum and proximal femur leading to subluxation or dislocation of the hip. It is usually noted at birth during the new born check with the Ortolani and Barlow tests that will reveal a clicking, dislocated or unstable hip. The incidence is 1 in 1000. There should be a high index of suspicion in patients with risk factors such as family history, breech position, female sex and oligohydramnios, and
should be followed up with specific hip examination and ultrasound scanning must be performed. Clinical examination may show positive Galeazzi test (affected thigh is shorter when both legs are held together with knees flexed) and decreased abduction on the dislocated side. Asymmetrical skin folds are a non-specific sign.
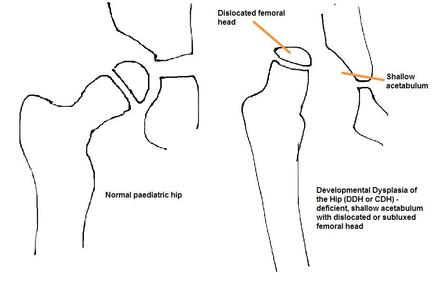
|
Early diagnosis and treatment of the hip leads to normal development and reduces the risk of early degeneration in the joint. A reducible hip in an infant less than 6 months old can be placed into a Pavlik harness for up to 12 weeks, holding the hip in a safe, reduced position and allowing it to mould to a more normal shape.
After 6 months of age, or if Pavlik treatment fails, patients will undergo gentle closed reduction and adductor/psoas tenotomies under general anaesthetic and application of a hip spica cast for 12 weeks. If that fails, at close to walking age, an open reduction with hip capsule tightening and femoral osteotomies with subsequent application of a hip spica cast for 12 weeks. Some patients with continued instability or particularly malformed hip joints may require further surgery later in life with proximal femoral osteotomies or pelvic osteotomies to achieve and maintain a stable reduction and allow for more normal hip development. Complications of DDH include avascular necrosis of the femoral head (often related to treatment), leg length discrepancy and early development of hip osteoarthritis. |
Legg-Calve-Perthes’ Disease (Perthes)
Perthes’ disease is due to disruption of the blood supply to the proximal femoral epiphysis leading to osteonecrosis followed by revascularisation and eventual remodelling. With osteonecrosis affecting the bone underlying the joint surface, there may be collapse, leading to the femoral head becoming malshaped and flattened. It is commonly seen in boys between 4-8 years of age with increased incidence with a positive family history. The incidence is 1 in 10,000. Patients will present with pain (sometimes knee pain) and a limp.
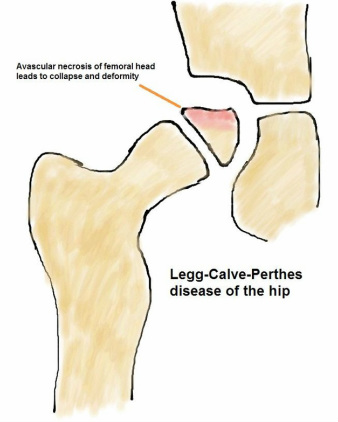
|
X-rays are diagnostic and prognosis is dependent on the amount of the femoral head involved and the age of the child (the older the child at presentation, the poorer the prognosis due to less time available for remodeling during growth). Management is aimed at controlling pain, protecting range of motion and maintaining the sphericity of the femoral head and rarely requires operation (such as proximal femoral or pelvic osteotomy). The long-term risk of Perthes’ disease is the development of early hip osteoarthritis. |
Slipped Upper Femoral Epiphysis (SUFE)
This is due to weakness in the proximal femoral physis leading to the femoral neck displacing anteriorly and rotating externally relative to the proximal femoral epiphysis. The incidence is 1 in 100,000. It is often seen in overweight adolescent boys aged 10-16 years old and is more common in Afro-Caribbean and Pacific Islander populations. Up to a quarter have both hips affected and slips in younger patients may be associated with
hormonal abnormalities (such as hypothyroidism). Examination reveals an antalgic (painful) gait, externally rotated leg and groin, thigh or knee pain.
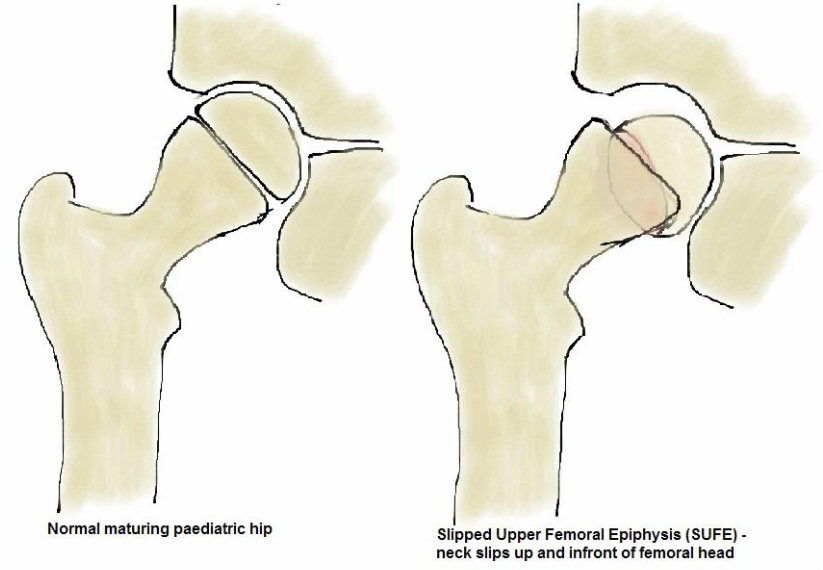
|
Slips may be acute or chronic and diagnosis is made on X-ray. Most slips require operative treatment with screws used to fix the epiphysis to the proximal femur and often the contralateral hip will be pinned prophylactically to prevent a slip. Complications include loss of the joint cartilage, avascular necrosis of the femoral head and early arthritis. |
Transient synovitis of the hip
Any child presenting with a painful limp must have septic arthritis, DDH, Perthes’ disease and SUFE ruled out with X-rays and blood tests (including inflammatory markers to rule out infection). Once these severe hip problems are excluded, the most likely diagnosis and most common cause of painful hip in childhood is transient synovitis. This is often related to a prior viral illness and the child presents non-weight bearing with limited painful hip motion. This is a self-limiting disorder and usually resolves over 24-48 hours, with anti-inflammatories used to ameliorate symptoms.
Osteomyelitis
Deep bone infection occurs most commonly in children in the metaphyses of long bones due to the extensive blood supply to these regions. Commonly caused by Staphylococcus aureus, haematogenous seeding of the bone occurs due to slow blood flow at the level of
the physis and metaphysis, leading to the formation of an abscess in the bone.
Clinically the child will be tender, warm and swollen in the region of the infection, usually with fever. X-rays and MRI are helpful in evaluating the area of bone affected. Blood tests will show elevated C-reactive protein (CRP), white blood cells and erythrocyte sedimentation rate (ESR) and blood cultures and image guided aspiration should be taken to confirm the diagnosis.
Treatment of an abscess is surgical drainage and debridement with administration of appropriate antibiotics. If detected early and if an abscess has not developed, antibiotics may be the only treatment required. Antibiotics are usually continued for 4-6 weeks, until the bone is quiescent, inflammatory markers have returned to normal and the child is clinically well.
Deep bone infection occurs most commonly in children in the metaphyses of long bones due to the extensive blood supply to these regions. Commonly caused by Staphylococcus aureus, haematogenous seeding of the bone occurs due to slow blood flow at the level of
the physis and metaphysis, leading to the formation of an abscess in the bone.
Clinically the child will be tender, warm and swollen in the region of the infection, usually with fever. X-rays and MRI are helpful in evaluating the area of bone affected. Blood tests will show elevated C-reactive protein (CRP), white blood cells and erythrocyte sedimentation rate (ESR) and blood cultures and image guided aspiration should be taken to confirm the diagnosis.
Treatment of an abscess is surgical drainage and debridement with administration of appropriate antibiotics. If detected early and if an abscess has not developed, antibiotics may be the only treatment required. Antibiotics are usually continued for 4-6 weeks, until the bone is quiescent, inflammatory markers have returned to normal and the child is clinically well.
Congenital talipes equinovarus (clubfoot)
This is a foot deformity more common in males that is due to shortened muscles and tendons (including the calf complex/Achilles tendon, tibialis posterior, flexor hallucis and flexor digitorum longus), joint capsules and ligaments around the foot resulting in forefoot adduction and supination with hindfoot equinus and varus. The incidence is 1 in 1000. Noticed at birth, the most common treatment is via the Ponseti method which usually
involves 6 weekly serial casts to improve the foot position and stretch the deforming structures. Often an Achilles tenotomy is also required. Following the casting, the patient has external rotation boots and bars applied for 2-3 years. Now that the Ponseti method is widely in use, surgical intervention is rare and is reserved for resistant, late presenting and stiff, syndromic clubfoot.
This is a foot deformity more common in males that is due to shortened muscles and tendons (including the calf complex/Achilles tendon, tibialis posterior, flexor hallucis and flexor digitorum longus), joint capsules and ligaments around the foot resulting in forefoot adduction and supination with hindfoot equinus and varus. The incidence is 1 in 1000. Noticed at birth, the most common treatment is via the Ponseti method which usually
involves 6 weekly serial casts to improve the foot position and stretch the deforming structures. Often an Achilles tenotomy is also required. Following the casting, the patient has external rotation boots and bars applied for 2-3 years. Now that the Ponseti method is widely in use, surgical intervention is rare and is reserved for resistant, late presenting and stiff, syndromic clubfoot.
